Input description¶
PCMSolver needs a number of input parameters at runtime. The API provides two ways of providing them:
by means of an additional input file, parsed by the
go_pcm.pyscript;by means of a special section in the host program input.
Method 1 is more flexible: all parameters that can be modified by the user are available. The host program needs only copy the additional input file to the scratch directory before execution. Method 2 just gives access to the core parameters.
In this page, input style and input parameters available in Method 1 will be documented.
Note that it is also possible to run the module standalone and use a classical charge
distribution.
The classical charge distribution can be specified by giving a molecular geometry
in the molecule section and an additional point multipoles distribution
in the charge distribution section.
The run_pcm executable has to be compiled for a standalone run with:
python <build-path/bin>/go_pcm.py --exe <build-path/bin> --inp molecule.inp
where the molecule.inp input file looks like:
units = angstrom
codata = 2002
medium
{
solvertype = cpcm
correction = 0.5
solvent = cyclohexane
}
cavity
{
type = gepol
area = 0.6
radiiset = uff
mode = implicit
}
molecule
{
# x, y, z, q
geometry = [0.000000000, 0.00000000, 0.08729478, 9.0,
0.000000000, 0.00000000, -1.64558444, 1.0]
}
The script and the executable do not need to be in the same directory.
Input style¶
The input for PCMSolver is parsed through the Getkw library written by Jonas Juselius
and is organized in sections and keywords. Input reading is
case-insensitive. An example input structure is shown below, there are also
some working examples in the directory examples. A general input
parameter has the following form (Keyword = [Data type]):
Units = [String]
CODATA = [Integer]
Cavity {
Type = [String]
NpzFile = [String]
Area = [Double]
Scaling = [Bool]
RadiiSet = [String]
MinRadius = [Double]
Mode = [String]
Atoms = [Array of Integers]
Radii = [Array of Doubles]
Spheres = [Array of Doubles]
}
Medium {
Nonequilibrium = [Bool]
Solvent = [String]
SolverType = [String]
MatrixSymm = [Bool]
Correction = [Double]
DiagonalIntegrator = [String]
DiagonalScaling = [Double]
ProbeRadius = [Double]
Green<GreenTag> {
Type = [String]
Der = [String]
Eps = [Double]
EpsDyn = [Double]
Eps1 = [Double]
EpsDyn1 = [Double]
Eps2 = [Double]
EpsDyn2 = [Double]
Center = [Double]
Width = [Double]
InterfaceOrigin = [Array of Doubles]
MaxL = [Integer]
}
}
Molecule {
MEP = [Bool]
Geometry = [Double]
}
ChargeDistribution {
Monopoles = [Double]
Dipoles = [Double]
}
MMFQ {
SitesPerFragment = [Integer]
Sites = [Array of Doubles]
NonPolarizable = [Bool]
}
Array-valued keywords will expect the array to be given in comma-separated format and enclosed in square brackets. The purpose of tags is to distinguish between cases in which multiple instances of the same kind of object can be managed by the program. There exist only certain legal tagnames and these are determined in the C++ code. Be aware that the input parsing script does not check the correctness of tags.
Input parameters¶
Available sections:
top section: sets up parameters affecting the module globally;
Cavity: sets up all information needed to form the cavity and discretize its surface;
Medium: sets up the solver to be used and the properties of the medium, i.e. the Green’s functions inside and outside the cavity;
Green, subsection of medium. Sets up the Green’s function inside and outside the cavity.
Molecule: molecular geometry to be used in a standalone run.
ChargeDistribution: sets up a classical multipolar (currently up to dipoles) charge distribution to use as additional source of electrostatic potential.
Note
The Molecule and ChargeDistribution sections only make sense in a standalone run,
i.e. when using the run_pcm executable.
Warning
Exactly matching results obtained from implementations of IEFPCM and/or CPCM (COSMO) given in other program packages requires careful selection of all the parameters involved. A partial checklist of parameters you should always keep in mind:
solvent permittivities (static and optical)
atomic radii set
scaling of the atomic radii
cavity surface
cavity partition (tesselation)
PCM matrix formation algorithm
strategy used to solve the PCM linear equations system.
Top section keywords¶
- Units
Units of measure used in the input file. If Angstrom is given, all relevant input parameters are first converted in au and subsequently parsed.
Type: string
Valid values: AU | Angstrom
Default: No Default
- CODATA
Set of fundamental physical constants to be used in the module.
Type: integer
Valid values: 2010 | 2006 | 2002 | 1998
Default: 2010
Cavity section keywords¶
- Type
The type of the cavity. Completely specifies type of molecular surface and its discretization. Only one type is allowed. Restart cavity will read the file specified by NpzFile keyword and create a GePol cavity from that.
Type: string
Valid values: GePol | Restart
Default: none
- NpzFile
The name of the
.npzfile to be used for the GePol cavity restart.Type: string
Default: empty string
- Area
Average area (weight) of the surface partition for the GePol cavity.
Type: double
Valid values: \(d \geq 0.01\,\text{a.u.}^2\)
Valid for: GePol cavity
Default value: \(0.3\,\text{a.u.}^2\)
- Scaling
If true, the radii for the spheres will be scaled by 1.2. For finer control on the scaling factor for each sphere, select explicit creation mode.
Type: bool
Valid for: all cavities except Restart
Default value: True
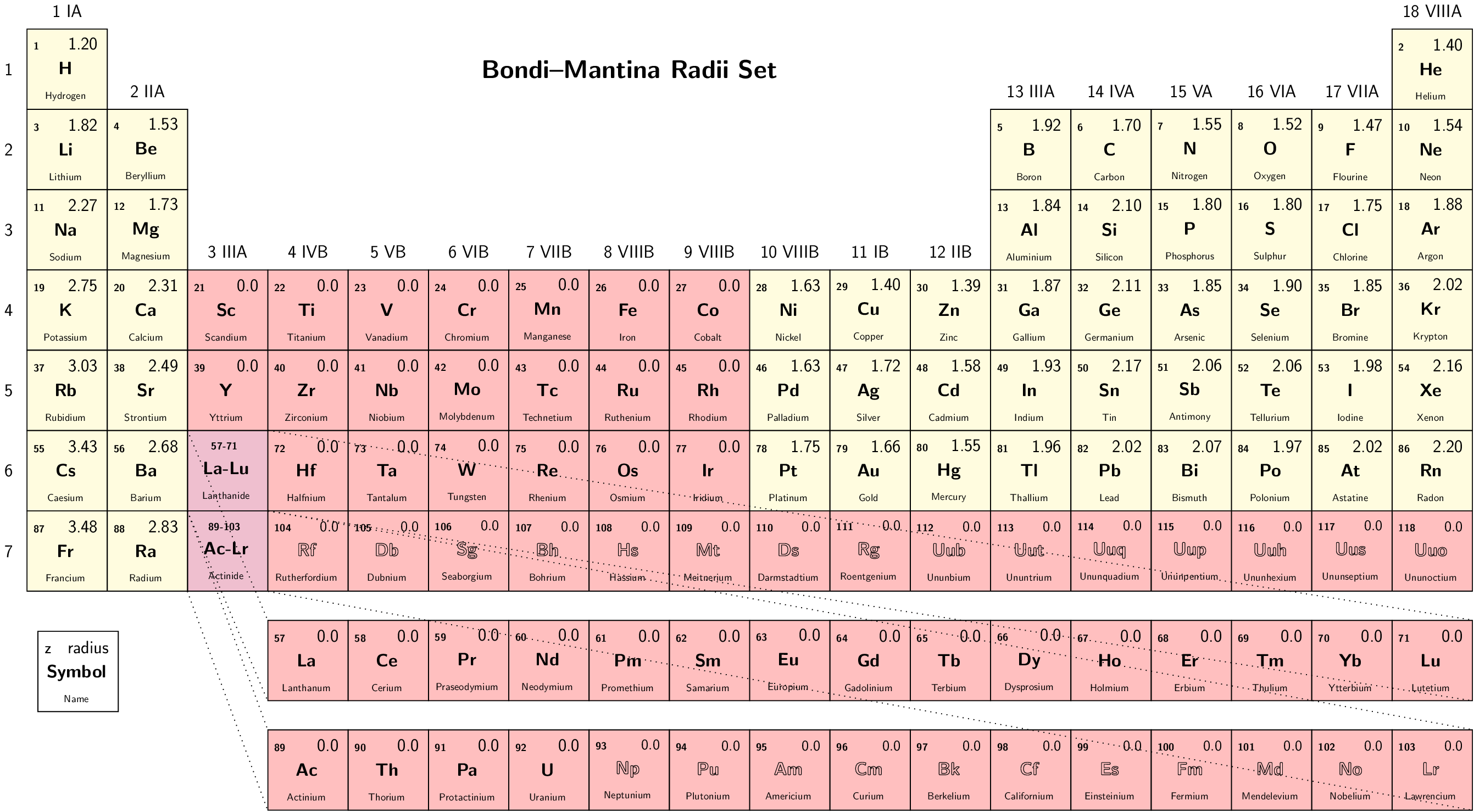
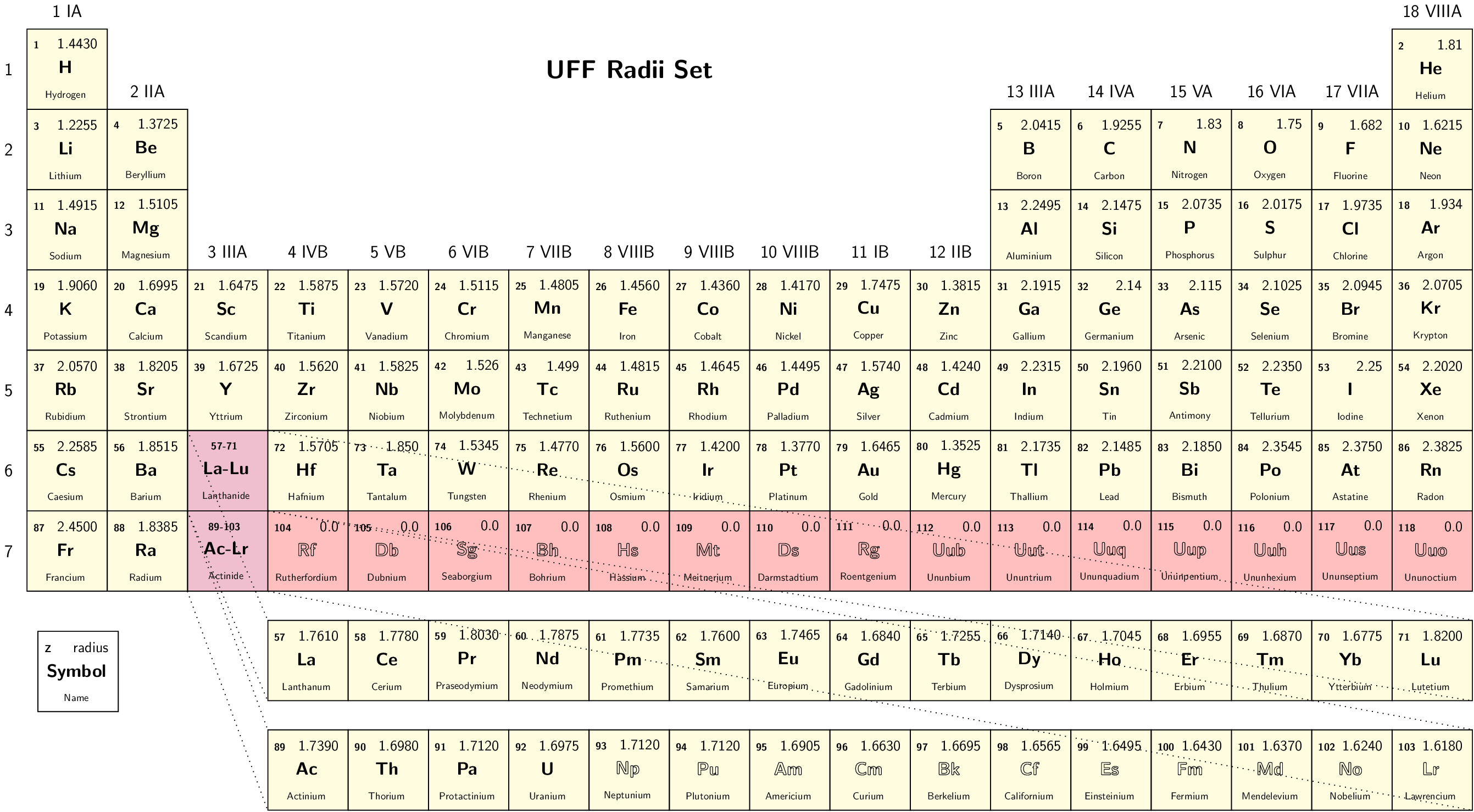
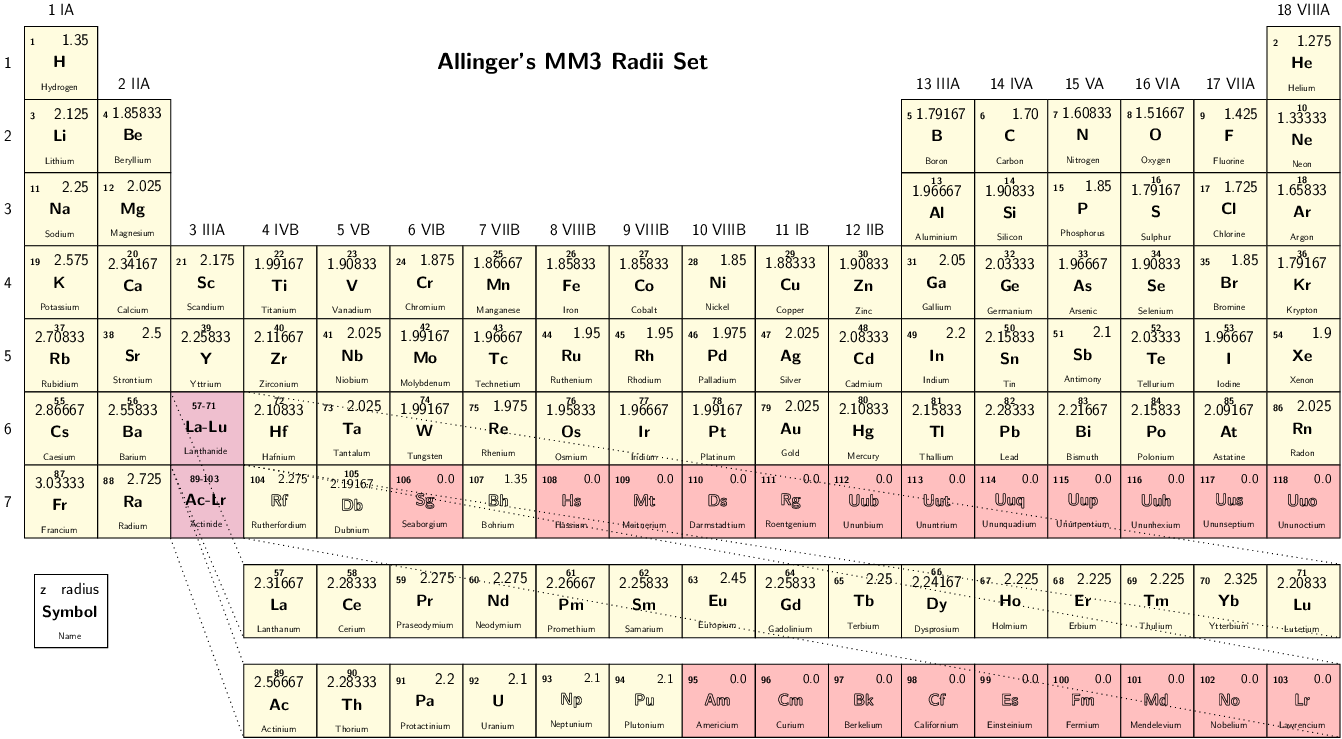
- RadiiSet
Select set of atomic radii to be used. Currently Bondi-Mantina [Bondi64][MantinaChamberlinValero+09], UFF [RCC+92] and Allinger’s MM3 [AZB94] sets available, see Available radii.
Type: string
Valid values: Bondi | UFF | Allinger
Valid for: all cavities except Restart
Default value: Bondi
Note
Radii in Allinger’s MM3 set are obtained by dividing the value in the original paper by 1.2, as done in the ADF COSMO implementation We advise to turn off scaling of the radii by 1.2 when using this set.
- MinRadius
Minimal radius for additional spheres not centered on atoms. An arbitrarily big value is equivalent to switching off the use of added spheres, which is the default.
Type: double
Valid values: \(d \geq 0.4\,\text{a.u.}\)
Valid for: GePol cavity
Default value: \(100.0\,\text{a.u.}\)
- Mode
How to create the list of spheres for the generation of the molecular surface:
in Implicit mode, the atomic coordinates and charges will be obtained from the QM host program. Spheres will be centered on the atoms and the atomic radii, as specified in one the built-in sets, will be used. Scaling by 1.2 will be applied according to the keyword Scaling;
in Atoms mode, the atomic coordinates and charges will be obtained from the QM host program. For the atoms specified by the array given in keyword Atoms, the built-in radii will be substituted by the radii provided in the keyword Radii. Scaling by 1.2 will be applied according to the keyword Scaling;
in Explicit mode, both centers and radii of the spheres are to be specified in the keyword Spheres. The user has full control over the generation of the list of spheres. Scaling by 1.2 is not applied, regardless of the value of the Scaling keyword.
Type: string
Valid values: Implicit | Atoms | Explicit
Valid for: all cavities except Restart
Default value: Implicit
- Atoms
Array of atoms whose radius has to be substituted by a custom value.
Type: array of integers
Valid for: all cavities except Restart
- Radii
Array of radii replacing the built-in values for the selected atoms.
Type: array of doubles
Valid for: all cavities except Restart
- Spheres
Array of coordinates and centers for construction of the list of spheres in explicit mode. Format is \([\ldots, x_i, y_i, z_i, R_i, \ldots]\)
Type: array of doubles
Valid for: all cavities except Restart
Medium section keywords¶
- SolverType
Type of solver to be used. All solvers are based on the Integral Equation Formulation of the Polarizable Continuum Model [CancesMennucci98]
IEFPCM. Collocation solver for a general dielectric medium
CPCM. Collocation solver for a conductor-like approximation to the dielectric medium
Type: string
Valid values: IEFPCM | CPCM
Default value: IEFPCM
- Nonequilibrium
Initializes an additional solver using the dynamic permittivity. To be used in response calculations.
Type: bool
Valid for: all solvers
Default value: False
- Solvent
Specification of the dielectric medium outside the cavity. This keyword must always be given a value. If the solvent name given is different from Explicit any other settings in the Green’s function section will be overridden by the built-in values for the solvent specified. See Table Available solvents for details.
Solvent = Explicit, triggers parsing of the Green’s function sections.Type: string
Valid values:
Water , H2O;
Propylene Carbonate , C4H6O3;
Dimethylsulfoxide , DMSO;
Nitromethane , CH3NO2;
Acetonitrile , CH3CN;
Methanol , CH3OH;
Ethanol , CH3CH2OH;
Acetone , C2H6CO;
1,2-Dichloroethane , C2H4CL2;
Methylenechloride , CH2CL2;
Tetrahydrofurane , THF;
Aniline , C6H5NH2;
Chlorobenzene , C6H5CL;
Chloroform , CHCL3;
Toluene , C6H5CH3;
1,4-Dioxane , C4H8O2;
Benzene , C6H6;
Carbon Tetrachloride , CCL4;
Cyclohexane , C6H12;
N-heptane , C7H16;
Explicit.
- MatrixSymm
If True, the PCM matrix obtained by the IEFPCM collocation solver is symmetrized \(\mathbf{K} := \frac{\mathbf{K} + \mathbf{K}^\dagger}{2}\)
Type: bool
Valid for: IEFPCM solver
Default: True
- Correction
Correction, \(k\) for the apparent surface charge scaling factor in the CPCM solver \(f(\varepsilon) = \frac{\varepsilon - 1}{\varepsilon + k}\)
Type: double
Valid values: \(k > 0.0\)
Valid for: CPCM solver
Default: 0.0
- DiagonalIntegrator
Type of integrator for the diagonal of the boundary integral operators
Type: string
Valid values: COLLOCATION
Valid for: IEFPCM, CPCM
Default: COLLOCATION
Notes: in future releases we will add PURISIMA and NUMERICAL as options
- DiagonalScaling
Scaling factor for diagonal of collocation matrices
Type: double
Valid values: \(f > 0.0\)
Valid for: IEFPCM, CPCM
Default: 1.07
Notes: values commonly used in the literature are 1.07 and 1.0694
- ProbeRadius
Radius of the spherical probe approximating a solvent molecule. Used for generating the solvent-excluded surface (SES) or an approximation of it. Overridden by the built-in value for the chosen solvent.
Type: double
Valid values: \(d \in [0.1, 100.0]\,\text{a.u.}\)
Valid for: all solvers
Default: 1.0
Green section keywords¶
If Solvent = Explicit, two Green’s functions sections must be specified
with tags inside and outside, i.e. Green<inside> and
Green<outside>. The Green’s function inside will always be the vacuum,
while the Green’s function outside might vary.
- Type
Which Green’s function characterizes the medium.
Type: string
Valid values: Vacuum | UniformDielectric | SphericalDiffuse | SphericalSharp
Default: Vacuum
- Der
How to calculate the directional derivatives of the Green’s function:
Numerical, perform numerical differentiation debug option;
Derivative, use automatic differentiation to get the directional derivative;
Gradient, use automatic differentiation to get the full gradient debug option;
Hessian, use automatic differentiation to get the full hessian debug option;
Type: string
Valid values: Numerical | Derivative | Gradient | Hessian
Default: Derivative
Note
The spherical diffuse Green’s function always uses numerical differentiation.
- Eps
Static dielectric permittivity of the medium
Type: double
Valid values: \(\varepsilon \geq 1.0\)
Default: 1.0
- EpsDyn
Dynamic dielectric permittivity of the medium
Type: double
Valid values: \(\varepsilon \geq 1.0\)
Default: 1.0
- Profile
Functional form of the dielectric profile
Type: string
Valid values: Tanh | Erf | Log
Valid for: SphericalDiffuse
Default: Log
- Eps1
Static dielectric permittivity inside the interface
Type: double
Valid values: \(\varepsilon \geq 1.0\)
Valid for: SphericalDiffuse, SphericalSharp
Default: 1.0
- EpsDyn1
Dynamic dielectric permittivity inside the interface
Type: double
Valid values: \(\varepsilon \geq 1.0\)
Valid for: SphericalDiffuse, SphericalSharp
Default: 1.0
- Eps2
Static dielectric permittivity outside the interface
Type: double
Valid values: \(\varepsilon \geq 1.0\)
Valid for: SphericalDiffuse, SphericalSharp
Default: 1.0
- EpsDyn2
Dynamic dielectric permittivity outside the interface
Type: double
Valid values: \(\varepsilon \geq 1.0\)
Valid for: SphericalDiffuse, SphericalSharp
Default: 1.0
- Center
Center of the interface layer. This corresponds to the radius of the spherical droplet.
Type: double
Valid for: SphericalDiffuse, SphericalSharp
Default: 100.0 a.u.
- Width
Physical width of the interface layer. This value is divided by 6.0 internally.
Type: double
Valid for: SphericalDiffuse
Default: 5.0 a.u.
Warning
Numerical instabilities may arise if a too small value is selected.
- InterfaceOrigin
Center of the spherical droplet
Type: array of doubles
Valid for: SphericalDiffuse, SphericalSharp
Default: \([0.0, 0.0, 0.0]\)
- MaxL
Maximum value of the angular momentum in the expansion of the Green’s function for the spherical diffuse Green’s function
Type: integer
Valid for: SphericalDiffuse, SphericalSharp
Default: 30
Molecule section keywords¶
It is possible to run the module standalone and use a classical charge
distribution as specified in this section of the input.
The run_pcm executable has to be compiled for a standalone run with:
python go_pcm.py -x molecule.inp
where the molecule.inp input file looks like:
units = angstrom
codata = 2002
medium
{
solvertype = cpcm
correction = 0.5
solvent = cyclohexane
}
cavity
{
type = gepol
area = 0.6
radiiset = uff
mode = implicit
}
molecule
{
# x, y, z, q
geometry = [0.000000000, 0.00000000, 0.08729478, 9.0,
0.000000000, 0.00000000, -1.64558444, 1.0]
}
- Geometry
Coordinates and charges of the molecular aggregate. Format is \([\ldots, x_i, y_i, z_i, Q_i, \ldots]\) Charges are always assumed to be in atomic units
Type: array of doubles
ChargeDistribution section keywords¶
Set a classical charge distribution, inside or outside the cavity No additional spheres will be generated.
- Monopoles
Array of point charges Format is \([\ldots, x_i, y_i, z_i, Q_i, \ldots]\)
Type: array of doubles
- Dipoles
Array of point dipoles. Format is \([\ldots, x_i, y_i, z_i, \mu_{x_i}, \mu_{y_i}, \mu_{z_i} \ldots]\) The dipole moment components are always read in atomic units.
Type: array of doubles
MMFQ section keywords¶
Set a classical fluctuating charge force field. This is incompatible with any options specifying a continuum model. No additional spheres will be generated.
- SitesPerFragment
Number of sites per MM fragment. For water this is 3.
Type: integer
Default: 3
- Sites
Array of MM sites for the FQ model Format is \([\ldots, x_i, y_i, z_i, chi_i, eta_i \ldots]\)
Type: array of doubles
- NonPolarizable
Whether to make this force field nonpolarizable.
Type: bool
Default: false
Available solvents¶
The macroscopic properties for the built-in list of solvents are:
static permittivity, \(\varepsilon_s\)
optical permittivity, \(\varepsilon_\infty\)
probe radius, \(r_\mathrm{probe}\) in Angstrom.
The following table summarizes the built-in solvents and their properties. Solvents are ordered by decreasing static permittivity.
Name
Formula
\(\varepsilon_s\)
\(\varepsilon_\infty\)
\(r_\mathrm{probe}\)
Water
H2O
78.39
1.776
1.385
Propylene Carbonate
C4H6O3
64.96
2.019
1.385
Dimethylsulfoxide
DMSO
46.7
2.179
2.455
Nitromethane
CH3NO2
38.20
1.904
2.155
Acetonitrile
CH3CN
36.64
1.806
2.155
Methanol
CH3OH
32.63
1.758
1.855
Ethanol
CH3CH2OH
24.55
1.847
2.180
Acetone
C2H6CO
20.7
1.841
2.38
1,2-Dichloroethane
C2H4Cl2
10.36
2.085
2.505
Methylenechloride
CH2Cl2
8.93
2.020
2.27
Tetrahydrofurane
THF
7.58
1.971
2.9
Aniline
C6H5NH2
6.89
2.506
2.80
Chlorobenzene
C6H5Cl
5.621
2.320
2.805
Chloroform
CHCl3
4.90
2.085
2.48
Toluene
C6H5CH3
2.379
2.232
2.82
1,4-Dioxane
C4H8O2
2.250
2.023
2.630
Benzene
C6H6
2.247
2.244
2.630
Carbon tetrachloride
CCl4
2.228
2.129
2.685
Cyclohexane
C6H12
2.023
2.028
2.815
N-heptane
C7H16
1.92
1.918
3.125